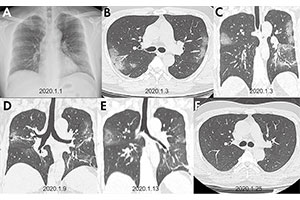
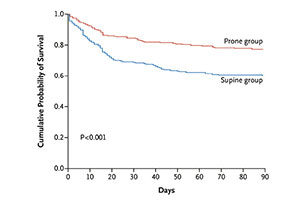
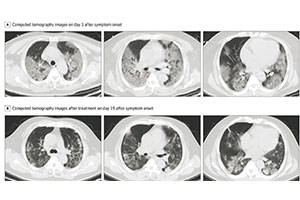
2009年,我在《生命新知》五月刊的卷首语上无厘头地写到:“在过去的300年间,共记录过6次流感世界大流行,每两次间隔时间为50年~60年,最近一次流感大流行发生在1968年。据此推算,下一次流感大流行的时间应在2021年之前”,此文章的标题是“必然的大流行”。
2020年,世界大流行终于要来了,虽然不是禽流感或猪流感,而是新型冠状病毒,也让我禁不住“哇”了一声:“我的预言很准”。但是,得意只是一瞬间,遂自责“乌鸦嘴”,汗毛竖起,细思恐极,尽管一切尽在规律之中:利用300年先验数据建立的模型及设计的算法,10年前预测了世界大流行的时间、模式、可能的地点、传播的路径,但人们被自大冲昏头脑,自以为是的态度及行为让人恐惧及悲哀。“没有”、“有限”、“较弱”定义敌人,“可控”、“一定”、“能力”认知自己,大流行应该会成真,病毒会染指地球村的每一个角落,无论你是村长、村委、会计、保安、小组长、村医、村民都无从幸免。
在2020大流行的前夜,让我们复读一下10年前的短文。
“自1975年以来, 世界上新发现了40多种人类致病微生物, 其中包括Ebola病毒、Nipah病毒、hanta病毒、SARS和HIV等。同时,现有的病原微生物也在不断变异,逃避着人类的免疫以及药物的杀伤。
虽然“聪明”的HIV仅有6个基因,但人类的免疫系统却成为其手下败将,全世界4000万人携带病毒,每年致300万人死亡,艾滋病已流行了25年;善变的SARS病毒完成了种系间的跳跃,启动超强的免疫反应,使人类死伤在自身免疫系统的火力之下,2003年导致大流行;顽固金葡菌在人类滥用抗生素的习惯下,变成了MRSA,并从医院蔓延到社区,造成了大流行;结核每年导致1000万人发病,多重耐药性结核已出现,同艾滋病狼狈为奸,正在流行;凶险的Ebola病毒已逃逸出非洲森林,东南亚的猪成为其新宿主,又向人类靠近了一大步。
如今,SARS 的阴影刚刚挥去,H5N1禽流感的恐惧还没有消减,猪流感又向人类袭来。一个月的时间,猪甲型H1N1 流感(swine influenza A,H1N1)病毒已流窜至世界四大洲。一片恐慌,甚至有人提议,将猪斩尽杀绝。
H1N1 本是禽流感病毒,在1918年大流行(pandemic)。当年3 月,美国堪萨斯州的兵营里出现士兵因流感死亡,至5月,全美国、欧洲、日本、中国均出现大流行,但死亡率不高。同年9月,第二个流行高潮到来,病程凶险,死亡率高达10%,导致全球5000万人死亡,其中,青壮年为主要死亡人群。之后,H1N1禽流感病毒在人体内不断减毒,每年于局部流行(epidemics),成为典型的人流感,与人类共生存。
因此,合理的科学推测是:人类将H1N1流感病毒传给了猪,与猪体内的病毒发生了基因重组,基因发生了变异,增加了其毒性,并跨越种系,感染了人类。因为人类对此变异病毒没有免疫力,同时,在感染变异病毒后产生剧烈的炎症反应,从而导致较高的死亡率。猪在此次事件中充当了一个“搅拌机”,而罪魁祸首可能还是我们人类。我们曾经将结核的罪名强加给牛,但近期研究表明,在人类饲养牛之前,牛没有患结核病的痕迹,是人将结核杆菌传染给了牛。人类对大自然的侵犯,对物种间平衡的破坏,让人类承受了沉重的惩罚。
在过去的300年间,共记录过6次流感世界大流行,每两次间隔时间为50年~60年,最近一次流感大流行发生在1968年。据此推算,下一次流感大流行的时间应在2021年之前。当今,世界人口是1918年的3.5 倍(60 亿),按20%的感染率,10%的死亡率计算,届时将有十数亿人患病,上亿人死亡,我们的公共卫生体系将无法应对。庆幸的是,本次流感的传染性及致死性不强,截至2009 年5 月6 日,全球共有1600 余人确诊为H1N1猪流感患者,死亡人数为44 人,死亡率不超过0.3%。人类要认真反思,精心准备,全球合作,阻止或延缓这次不可避免的流感大流行。但历史的经验表明,需要警惕H1N1继续变异、毒性增强,预防可能在秋天出现的第二波袭击。”
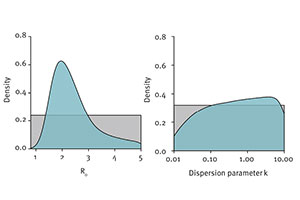
借用十年前的卷首语,不是偷懒,是面对新冠病毒疫情的全球走向、趋势、态度、举措,实在无语。尽管如此,还是要感慨几句:新冠病毒不是流感,目前的数据显示,她(R0=2.6)比流感(R0=1.3)传染力强两倍,潜伏期长三倍。好多未知,希望权威们不要太快下结论,要学会说“不知道”、“不确定”。疫苗不会在2个月内造出来;新药要经过严格的研究和验证,方具备写入治疗指南的基础;政客不是科学家,不能用无科学依据的行政命令、哲学、推理、传统治病方式防疫;数万名医生护士从全国驰援武汉,再次成为中国最可爱的人,再次被贴上天使的标签,但愿疫情过后不要健忘得太快。
请关注本期疫情复盘专刊。